

Hopefully, some of you have been following my series on baraminology that has been posting periodically throughout the last few months. In this article, I’m going to take time off from working on those articles and demonstrate how to use the BLAST software available from the NIH, discuss how this technology applies to baraminology, and give you a brief sneak peek as to what is coming in the tenth baraminology article. Learning how to use this BLAST technology has been a blast and I hope this brief tutorial is helpful to all budding baraminologists. Warning, this tutorial assumes that you have a basic understanding of genetics.
The BLAST technology takes a database of mitochondrial, whole genomic, and RNA sequences and makes them available to researchers for comparison. The acronym BLAST stands for Basic Local Alignment Search Tool. Essentially, the tool allows scientists and amateurs alike to use the publically available DNA and RNA sequences to determine how similar they are. The tool also allows for comparison on protein sequences and transcribing between proteins and RNA going both directions.
In order to use the BLAST tool, you will need access to two websites. The first is the National Center for Biotechnology Information website, which houses the database of genomic sequences. You will also need access to the BLAST tool. Once you have both of those open, you will need to select Nucleotide Blast to get started using the genome comparer. For this example, we will compare the whole genomes of the Green anole (Anolis carolinensis) with that of the cocoa tree (Theobroma cacao cultivar). The comparison is deliberately ridiculous.
Once you have decided what organisms to compare, you will need to open the BLAST tool. When you do, and have selected nucleotide BLAST, you will be greeted with the below screen.
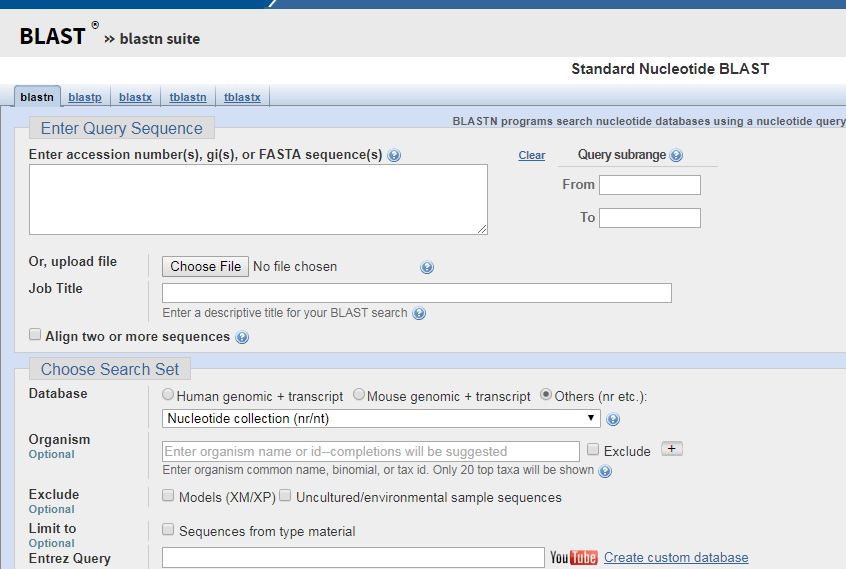
It looks intimidating but its a lot easier to use than it seems. To compare multiple sequences, select the box marked “Align two or more sequences”. This will bring up a new screen.
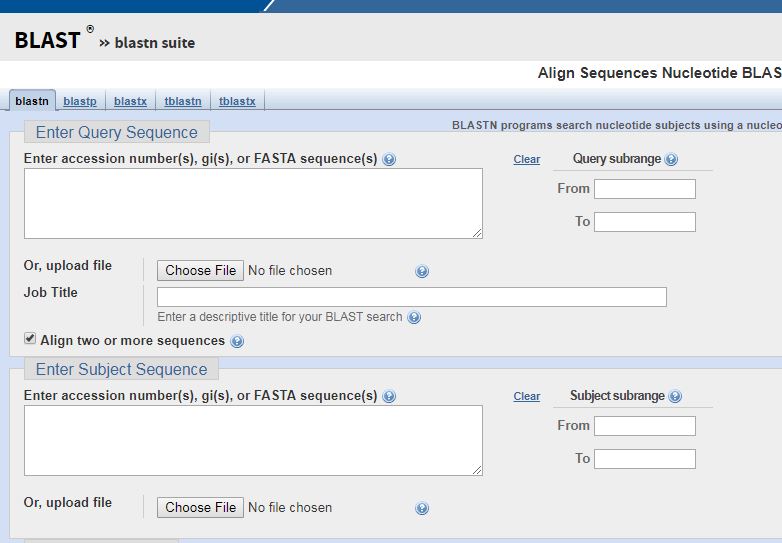
Once on this screen, you’re almost ready to begin comparing. All you need now is sequences to compare. In order to compare them, you will need to search the database provided above for a sequence you want to compare. To find the sequence, simply type what you’re looking for into the database search engine. With a little practice, you can generally find what you’re looking for fairly easily. In this case, the first search was for the genome of the green anole. However, the green anole genome is too big for one file. It is broken down by chromosome.
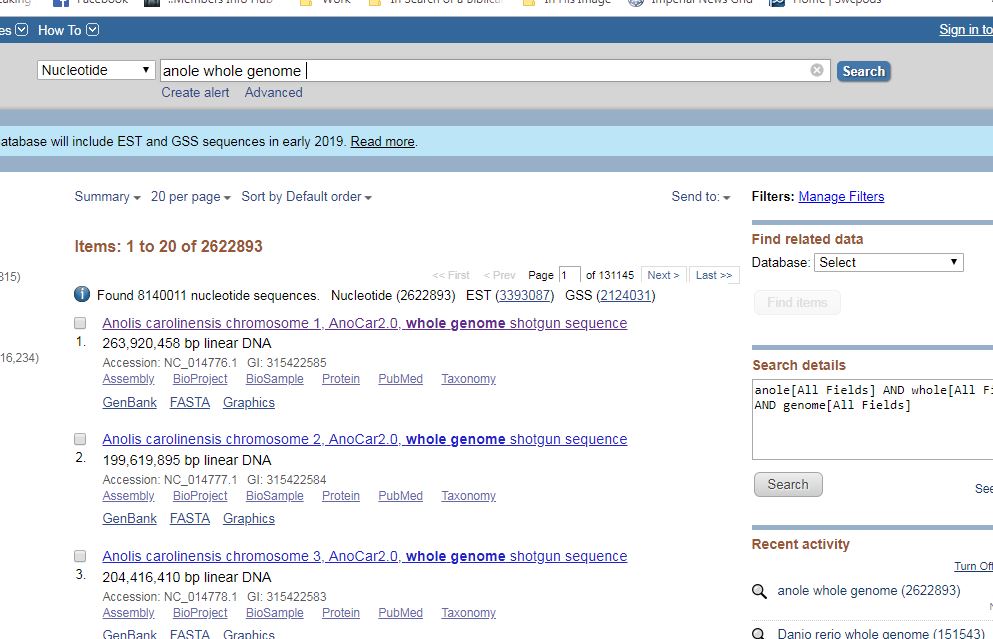 You use the same process to locate the cocoa tree genome. Since it is a bit smaller, we will be comparing the first six chromosomes of the cocoa tree to the first chromosome of the anole. Yes it’s absurd, but this is a tutorial so stay with me.
You use the same process to locate the cocoa tree genome. Since it is a bit smaller, we will be comparing the first six chromosomes of the cocoa tree to the first chromosome of the anole. Yes it’s absurd, but this is a tutorial so stay with me.
In order to compare sequences, you will need to place either the accession number or the GI number of each sequence you wish to compare in one of the two spots on the BLAST software. You can see these numbers directly below the listing of the number of base pairs in each sequence. Once you’re ready, click the BLAST button at the bottom of the screen and it will begin comparing. It may take some time with sequences that are lengthy, like the ones we selected for this tutorial. For shorter sequences, it will be instantaneous. In this case, running this query yielded no significant similarity, unsurprisingly.
However, using smaller sequences, each of mammals, yields different results. Comparing the mtDNA sequences of the walrus (Odobenus rosmarus rosmarus) and the elephant seal (Mirounga leonina) yields the below results.
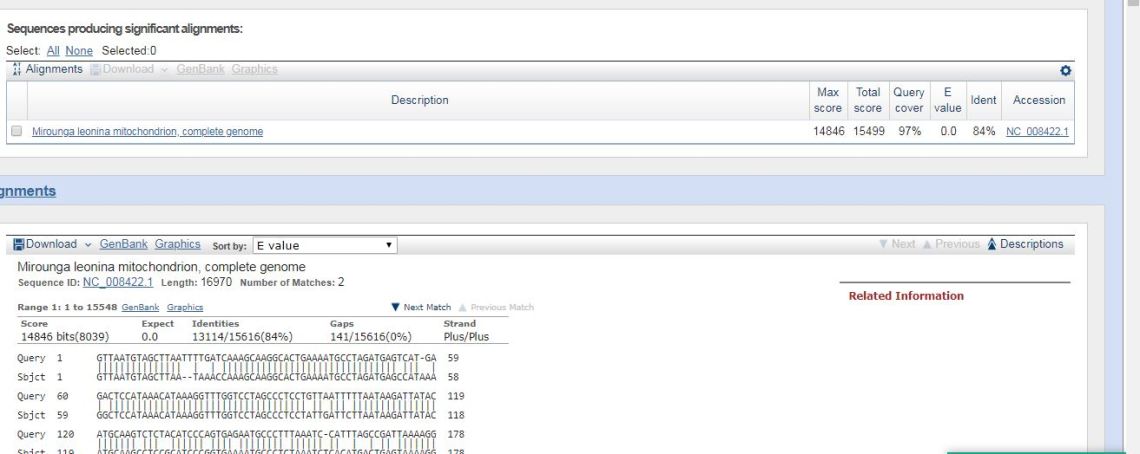
In case you can’t read that, mtDNA sequencing results in an 84% similarity. By contrast, humans and chimps, using the same methodology, have 92% similarity. This is why mtDNA is not the arbiter of the baramins.
Instead, individual whole genomes, and or sequences need to be used to try to discern similarity. Using genetic data should only be used when the only other alternative is statistics. If forced to use individual genes, use as many genes as you can and average the percentage differences to get an average similarity of the genome.
Hopefully, this simple, short tutorial on how to use BLAST, as well as how to interpret its results, will help the amateur baraminologist make more accurate assessments of the natural world. Hopefully, it will also demystify the use of genetic data in both baraminology, and the human/chimp comparison argument.
